




Forschungsgruppe Stegmüller
Gene und molekulare Mechanismen bei motorischen Erkrankungen
Im meinem Labor gilt das Forschungsinteresse Genen und molekularen Programmen, die in Zusammenhang mit motorischen Erkrankungen stehen. Wir nutzen molekulare und biochemische Techniken zusammen mit Mausgenetik und Verhaltensanalysen, um Gene zu untersuchen, die in Entwicklungsstörungen und degenerativen Erkrankungen des Gehirns wie z.B. Ataxien und Parkinsonismus eine Rolle spielen. Eine zunehmende Anzahl an Studien weist auf die Bedeutung des Ubiquitin Proteasom Systems (UPS) in diesen Erkrankungen hin. Das UPS ist einerseits für die Beseitigung von ubiquitinierten Proteinen verantwortlich, andererseits ist die Ubiquitinierungen von Proteinen auch für Signalkaskaden in der Zelle wichtig. Daher sind wir daran interessiert wie das UPS an der Gehirnentwicklung und –degeneration beteiligt ist.
Leitung
PD Dr. rer. nat. Judith Stegmüller
Tel. +49 - 241 - 8085793
Fax. +49 – 241 – 80 82582
jstegmuellerukaachende

Rosanna Huchzemeier (BSc student)
Sabitha Joseph (Postdoctoral Fellow)
Daniel Komnig (Postdoctoral Fellow)
Yuhao Huang (Doctoral Student)
Quan Wang (Doctoral Student)
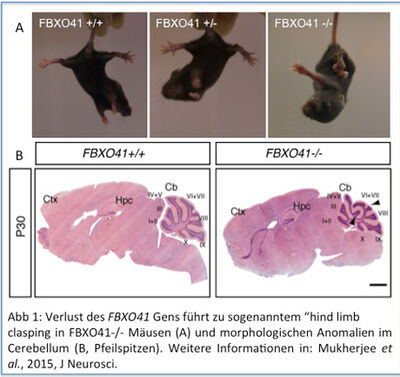
In einem unserer Projekte untersuchen wir die Funktion des UPS in der Kleinhirnentwicklung und damit molekulare Grundlagen für Ataxien. Wir haben eine transgene Mauslinie hergestellt, in der das FBXO41 Gen entfernt wurde. Die Mäuse sind durch ataxische Bewegungen und verzögerte, neuronale Migration im postnatalen Cerebellum charakterisiert. Des Weiteren haben wir Neurodegeneration im adulten Cerebellum beobachtet (Abb 1), 1. In weiteren Studien werden wir die Signalwege untersuchen, die von der ZNS-spezifischen E3 Ligase FBXO41 reguliert werden und die Rolle dieser Signalwege in Ataxien bestimmen.
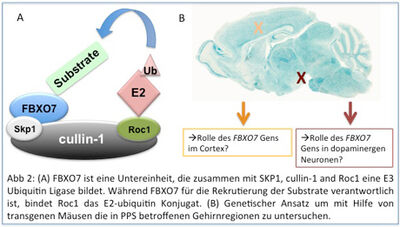
Ein anderes Projekt beschäftigt sich mit dem PARK15 Gen, das mit einer juvenilen Form von Parkinsonismus, dem sogenannten Parkinsonian-Pyramidal Syndrom (PPS) assoziiert wurde 2-6. PARK15 kodiert für die E3 Ligase FBXO7, deren Funktion in Neuronen wenig erforscht ist. Unsere Ziele sind zum Einen die Relevanz von FBXO7 im Gehirn mit Hilfe von genetisch veränderten Mäusen zu klären und zum Anderen die zellulären System zu identifizieren, die durch FBXO7 kontrolliert werden (Abb 2).
Mukherjee C, Holubowska A, Schwedhelm-Domeyer N et al. Loss of the Neuron-Specific F-Box Protein FBXO41 Models an Ataxia-Like Phenotype in Mice with Neuronal Migration Defects and Degeneration in the Cerebellum. J Neurosci 2015; 35:8701-8717.
Di Fonzo A, Dekker MC, Montagna P et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 2009; 72:240-245.
Yalcin-Cakmakli G, Olgiati S, Quadri M et al. A new Turkish family with homozygous FBXO7 truncating mutation and juvenile atypical parkinsonism. Parkinsonism Relat Disord 2014; 20:1248-1252.
Shojaee S, Sina F, Banihosseini SS et al. Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet 2008; 82:1375-1384.
Paisan-Ruiz C, Guevara R, Federoff M et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord 2010.
Lohmann E, Coquel AS, Honore A et al. A new F-box protein 7 gene mutation causing typical Parkinson's disease. Mov Disord 2015; 30:1130-1133.