






Forschung am Zentrum für Humangenetik und Genommedizin
Neben der Lehre und Krankenversorgung ist uns als universitäre Einrichtung die Erforschung von genetischen Erkrankungen ein besonderes Anliegen.
Neue Technologien und verbesserte Rechenleistungen haben die Forschung im Bereich der Humangenetik in den letzten Jahren revolutioniert. Je nach Fragestellung nutzen wir die verschiedenen Techniken der Hochdurchsatzsequenzierung, um das molekulare Verständnis menschlicher Erkrankungen zu erforschen:
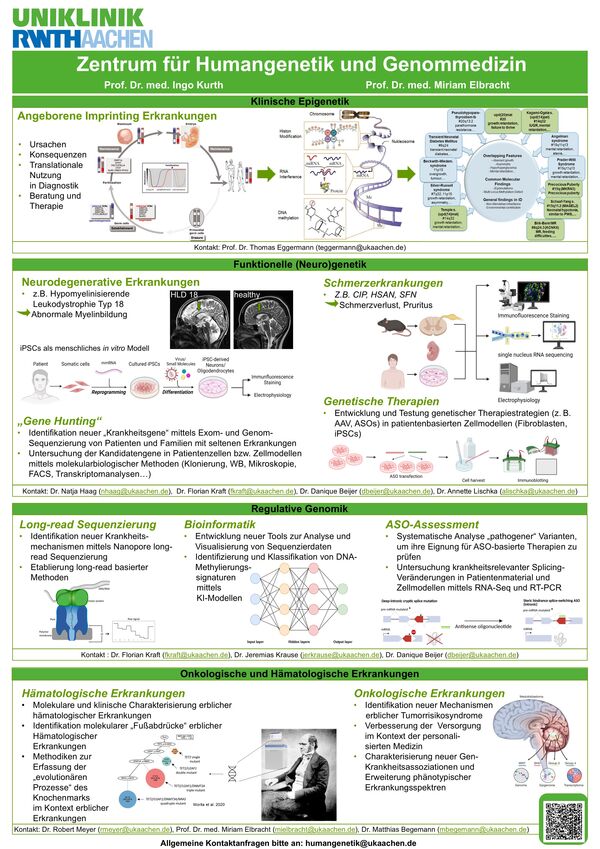
Zu unseren thematischen Schwerpunkten im Bereich der Grundlagenforschung und Genommedizin gehören die Erforschung von
- Schmerzerkrankungen
- Imprintingerkrankungen
- Data Science and Machine Learning in Genomics
- erblichen Tumorerkrankungen
- sowie die translationale Forschung im Bereich der Klinischen Genomik
Dank der Zusammenarbeit mit anderen Forscherinnen und Forschern sowie der Unterstützung durch Fördermittel können wir unsere Forschung auf höchstem Niveau betreiben.
Forschungsfragen im Bereich Genomik werden auch in Zusammenarbeit mit der IZKF Genomics Facility bearbeitet.
| Sollten Sie Interesse an einer Dissertation oder Masterarbeit haben, entnehmen Sie die Informationen hierzu bitte der Seite Dissertationen und Masterarbeiten. |
|---|