

Myelofibrosis and MPN
Under physiologic conditions, the bone marrow (BM) is composed of a sophisticated network of fibers and cells, which together provide the fertile soil for coordinated and demand-adapted blood cell production. Bone marrow fibrosis (myelofibrosis, MF) is a disorder in which normal BM tissue and blood-forming cells are gradually replaced by prominent coarse fibers and scar-like tissue. Over time, this eventually leads to failure of the BM to produce blood cells and translocation of blood-forming tissues to extramedullary, ontogenetically early hematopoietic organs such as spleen and liver. Together, this results in hematopoietic insufficiency, (hepato-)splenomegaly, and impaired survival. Polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) constitute the classical Bcr-Abl-negative myeloproliferative neoplasms (MPN) and chronic myeloid leukemia (CML) represents the unique Bcr-Abl MPN, all of which are clonal disorders of hematopoiesis, arising in the hematopoietic stem cell (HSC) compartment.
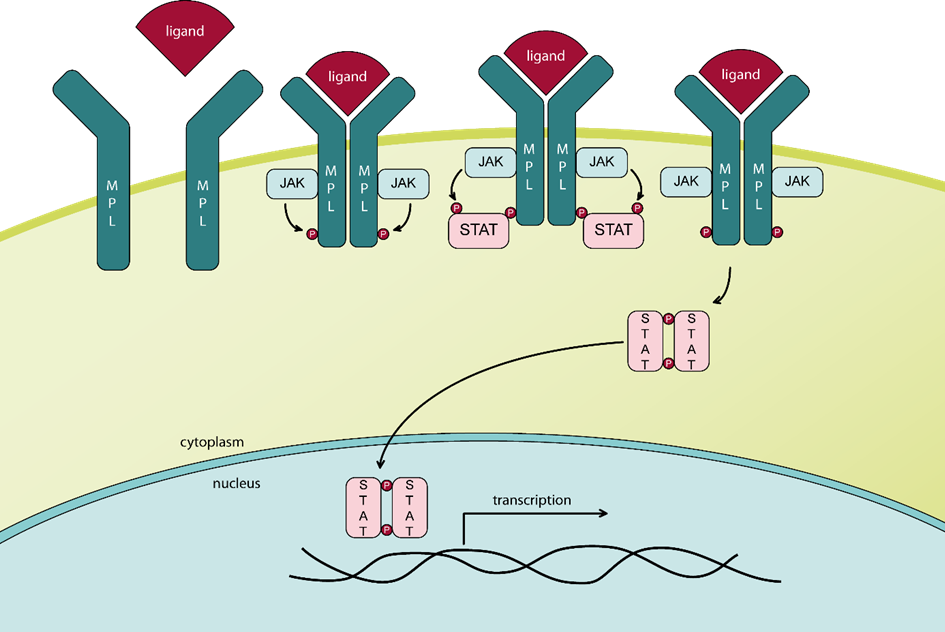
Almost all patients with BCR-ABL negative MPN harbor typical “driver” mutations in genes that either directly or indirectly activate JAK–STAT signaling. These mutations are located in the genes JAK2 (PV, ET, PMF), calreticulin (ET, PMF), or MPL (ET, PMF). 60% of patients with PMF harbor the JAK2V617F mutation, approximately 30% carry a calreticulin mutation (CALR), and 8% carry a myeloproliferative leukemia virus oncogene (MPL) mutation. Additional gene mutations (e.g. ASXL1, EZH2, IDH1, IDH2, SRSF2) are frequent. PMF is the least common of the three classic Bcr-Abl negative MPN; however, it is the most aggressive and is associated with a significantly increased morbidity and shortened survival. PMF is characterized by malignant clonal hematopoiesis, bone marrow fibrosis, extramedullary hematopoiesis, splenomegaly, and abnormal cytokine expression, leading to significant systemic symptoms and an elevated risk of thrombosis, severe bleeding, and transformation to acute leukemia.
CML is characterized by the presence of the BCR-ABL oncogene, which directly transforms hematopoietic stem and progenitor cells, enhancing their granulopoietic potential, proliferation, and survival. CML patients exhibit constitutional symptoms, splenomegaly, neutrophilia and leukocytosis, and, without treatment, their survival is dismal. Fortunately, most patients can be successfully treated with BCR-ABL inhibitors and achieve molecular remission. However, several factors, including BM fibrosis, can impede a favorable treatment response, putting these CML patients at risk for progression to acute leukemia, which carries a poor prognosis.
Driver mutations in MPN
Most BCR-ABL negative MPN patients harbor one of three mutually exclusive driving mutations causing the respective disease. 60% of patients with PMF harbor the JAK2V617F mutation, approximately 30% carry a calreticulin mutation (CALR), and 8% carry a myeloproliferative leukemia virus oncogene (MPL) mutation. The mechanisms, by which these alter cell function and survival, are outlined below.
MPLW515 mut
Myeloproliferative leukaemia protein (MPL), also termed thrombopoietin receptor, is a growth factor receptor encoded by the MPL proto-oncogene. The most frequent MPL mutation in the MPN context is a G to T transversion in exon 10 [1]. The resulting MPLW515L variant of the receptor is constitutively activated, leading to aberrant JAK-STAT signalling [2].
JAK2V617F exon 14 and JAK2 exon 12 mutations
The Janus kinase 2, encoded by the JAK2 gene, is a non-receptor tyrosine kinase responsible for the phosphorylation of MPL upon ligand-induced dimerization and of STAT5 [3], thereby activating STAT signalling.
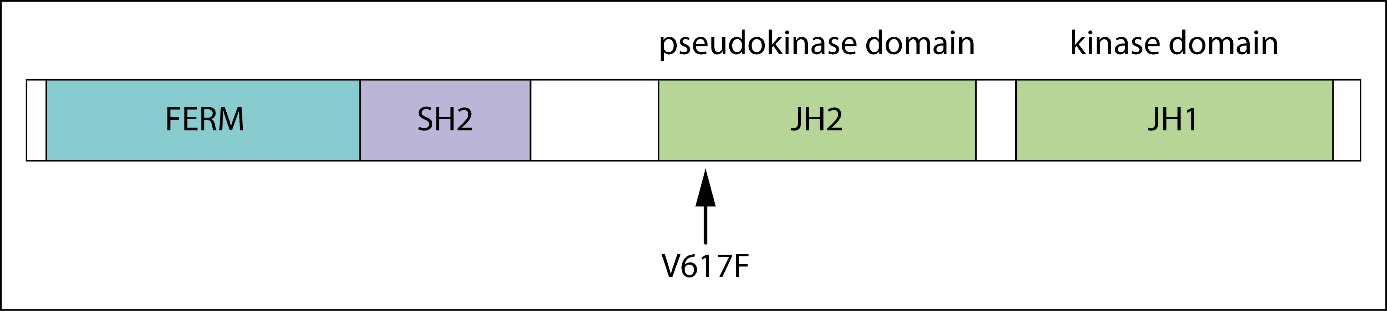
The figure above shows the functional domains of JAK2 and indicates the locus of the V617F substitution in the JH2 domain [4]. The basal kinase activity of the JH1 domain of JAK2 is autoinhibited by its JH2 domain in healthy cells [5]. In patients harboring the JAK2V617F mutation, which is defined by a G to T exchange in exon 14 of the JAK2 gene [4, 6], the interaction between JH1 and JH2 is disrupted, allowing the JH1 kinase domain to remain active [7]. This results in altered JAK-STAT signalling and is the most frequent cause for the development of a classical MPN [6]. Alternatively, mutations in exon 12 of JAK2 may be found, and these are typically associated with PV (rubra).
CALRDEL52 and CALRINS5
Calreticulin (CALR) is a chaperone for glycoproteins as well as a calcium buffer in the endoplasmic reticulum (ER) compartment of the cell. It is encoded by the CALR gene. The most frequently occurring mutations in CALR have been categorized as type I and type II mutations, wherein type I mutations are characterized by a 52 bp-deletion and type II mutations by a 5 bp-insertion in exon 9 of the CALR gene [8]. Both mutations result in a +1 bp frameshift [9] creating a common novel C-terminus that no longer includes an ER retention signal [10], allowing CALR to interact with proteins outside of the ER lumen.

The figure above displays the C-terminal amino acid sequence in CALR wildtype (CALRWT), 52 bp deletion (CALRDEL52) and 5 bp-insertion (CALRINS5) variants. It has been shown that the common C‑terminus of mutated CALR is essential for its oncogenic capability [10, 11]. Furthermore, the CALRMUT C‑terminus seems to bind the thrombopoietin receptor MPL and induce MPL homo-dimerization through mechanisms largely unknown [12], constitutively activating MPL and thereby the JAK-STAT signalling pathway [13].
BCR-ABL
The BCR-ABL oncogene is a fusion gene resulting from a reciprocal translocation between chromosome 9 and chromosome 22 (Philadelphia chromosome) first described in 1960 [14, 15].
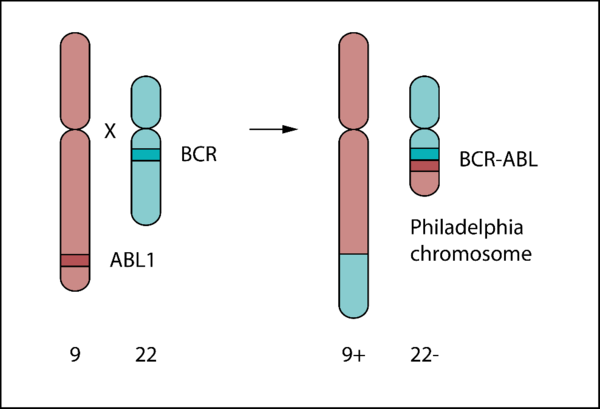
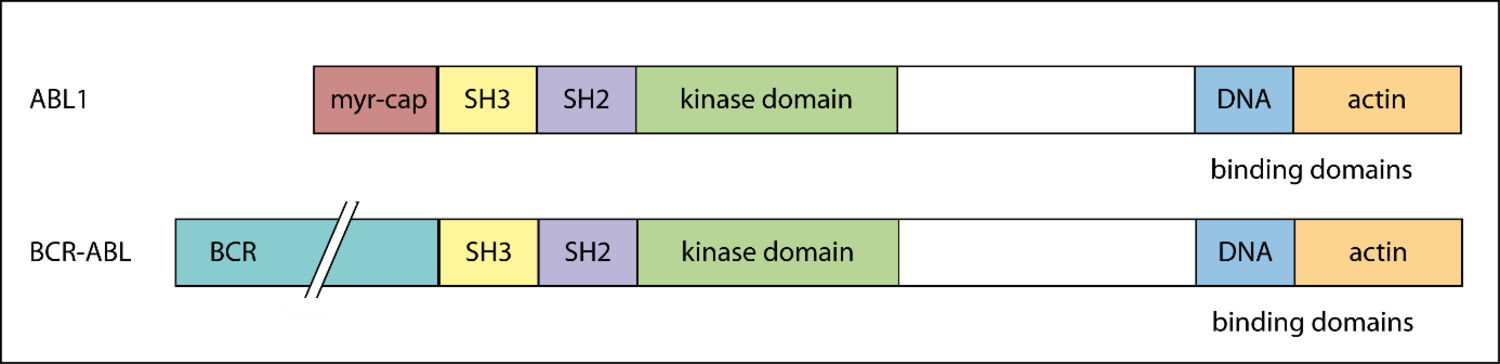
Physiological ABL1 codes for a tyrosine kinase involved in cell proliferation, differentiation, and survival. The fusion protein lacks an N-terminal myristoylated cap, undermining autoinhibitory SH2 and SH3 docking mechanisms and rendering BCR-ABL constitutively active [16, 17].
[1] Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010
[2] Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006
[3] Fujitani, Y., Hibi, M., Fukada, T. et al. An alternative pathway for STAT activation that is mediated by the direct interaction between JAK and STAT. Oncogene. 1997
[4] Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005
[5] Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002
[6] Jatiani SS, Baker SJ, Silverman LR, Reddy EP. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: approaches for targeted therapies. Genes Cancer. 2010
[7] Lee TS, Ma W, Zhang X, Giles F, Kantarjian H, Albitar M. Mechanisms of constitutive activation of Janus kinase 2-V617F revealed at the atomic level through molecular dynamics simulations. Cancer. 2009
[8] Ayalew Tefferi, Emnet A. Wassie, et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. American journal of hematology. 2014
[9] Misa Imai, Marito Araki, and Norio Komatsu. Somatic mutations of calreticulin in myeloproliferative neoplasms. International journal of hematology. 2017
[10] Shannon Elf, Nouran S. Abdelfattah, et al. Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer discovery. 2016
[11] Han L, et al. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J Hematol Oncol. 2016
[12] Shannon Elf, Nouran S. Abdelfattah, April J. Baral, Ann Mullally, Edwin Chen, and Danielle Beeson. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood. 2017
[13] Majed Dasouki, Irfan Saadi, and Syed O. Ahmed. THPO-MPL pathway and bone marrow failure. Hematology/oncology and stem cell therapy. 2015
[14] Nowell P, Hungerford D. Chromosome Studies on Normal and Leukemic Human Leukocytes. J Natl Cancer Inst. 1960
[15] Nowell P, Hungerford D. A minute chromosome in human chronic granulocytic leukemia. Science. 1960
[16] Franz WM, Berger P, Wang JY. Deletion of an N-terminal regulatory domain of the c-abl tyrosine kinase activates its oncogenic potential. EMBO J. 1989
[17] Nagar B, Hantschel O, Young MA, et al. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 2003