Genetisch bedingte Stoffwechselerkrankungen sind seltene und in ihrem Verlauf meist schwere, fortschreitende und lebensverkürzende Krankheiten. Die meist jungen Betroffenen leiden häufig an motorischen und neurologischen Symptomen, die nur bedingt therapierbar sind. In einer aktuellen Studie unter Leitung von Prof. Dr. Ingo Kurth, Direktor des Instituts für Humangenetik an der Uniklinik RWTH Aachen, und Prof. Dr. Thorsten Hornemann vom Institut für Klinische Chemie am Universitätsspital Zürich konnte erstmals das DEGS1-Gen als ursächlich für eine Erkrankung mit gestörtem Lipidmetabolismus identifiziert werden. Eine Sphingolipid-reiche Diät könnte einen ersten Ansatzpunkt für die Therapie dieser seltenen Erkrankung darstellen.
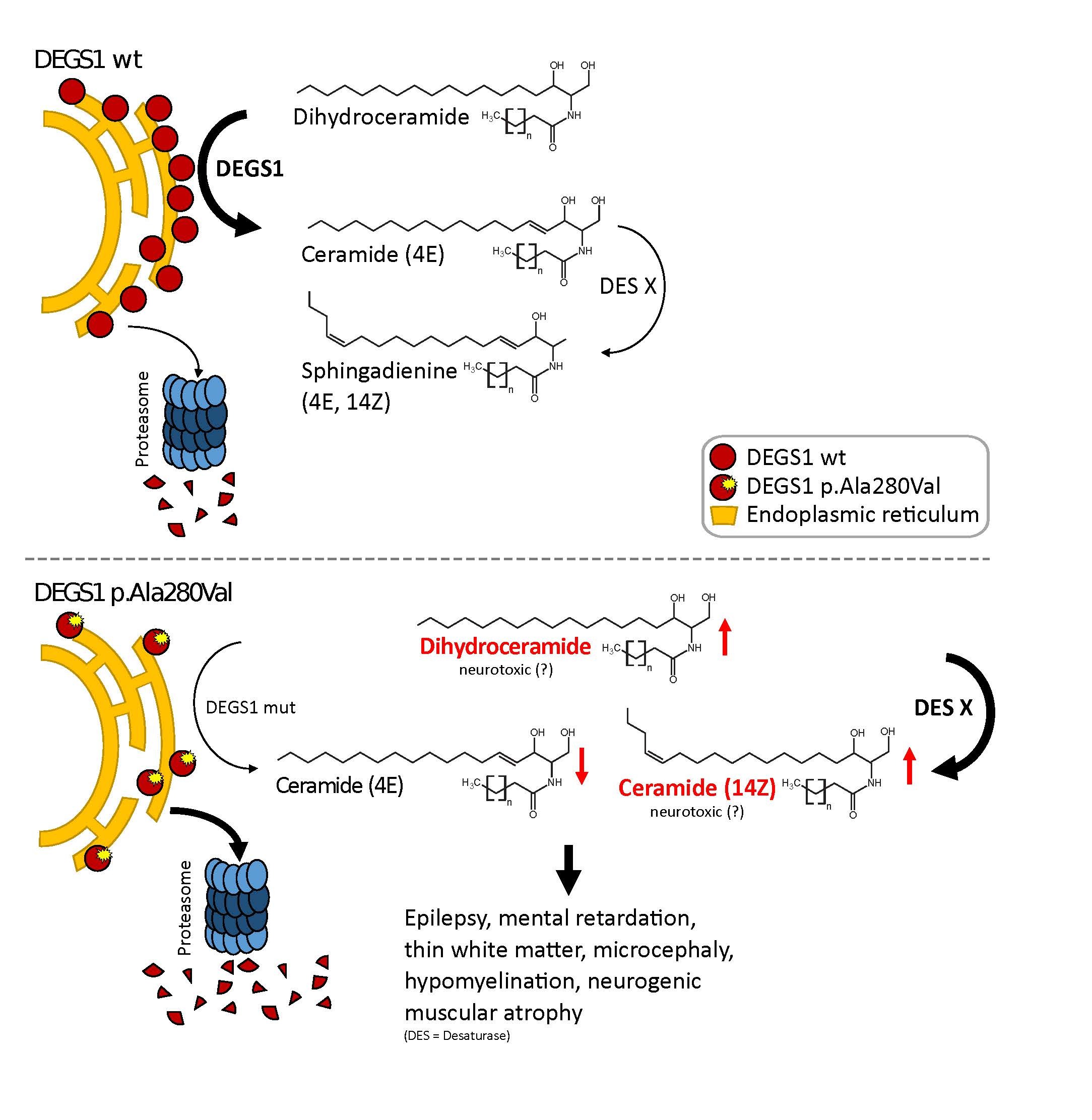
Sphingolipide sind wichtige Bestandteile der Zellmembranen und von besonderer Bedeutung für das Nervensystem, wo sie eine wichtige Rolle bei der Umhüllung von Nervenzellfortsätzen und der Übertragung von Nervenimpulsen spielen. Erkrankungen des Sphingolipid-Stoffwechsels können alle Organe betreffen, manifestieren sich aber oft am Nerven. Betroffene Patienten leiden durch den fortschreitenden Untergang von Zellen des zentralen und peripheren Nervensystems unter anderem anBewegungsstörungen, Krampfanfällen und kognitiven Einschränkungen.
Im Rahmen einer sogenannten Trio-Exom-Sequenzierung, bei der die Genome von Patienten und deren Eltern untersucht und miteinander verglichen werden, konnten die Forscher in Kombination mit einer neuen Sequenziertechnologie, der sogenannten Nanopore-Sequenzierung, eine genetische Veränderung im DEGS1-Gen als Ursache einer Erkrankung des Nervensystems identifizieren. Das betroffene Genprodukt, die Dihydroceramid-Desaturase 1 (DEGS1), ist ein Enzym des Sphingolipidmetabolismus und wichtig für die Synthese von Ceramiden. In Zusammenarbeit mit der Forschungsgruppe um Prof. Dr. Thorsten Hornemann konnten anschließend im Blut sowie in kultivierten Hautbiopsie-Zellen einer betroffenen Person durch Lipidomics-Analysen veränderte Sphingolipid-Spiegel detektiert werden. Neben einer reduzierten Proteinstabilität, welche eine verminderte Aktivität des DEGS1-Enzyms zur Folge hatte, konnte ein bisher unbekanntes und möglicherweise neurotoxisches Nebenprodukt des gestörten Stoffwechselweges als Ursache der Erkrankung nachgewiesen werden. Die Publikation befindet sich im Journal of Clinical Investigation, einer renommierten Fachzeitschrift für klinisch-angewandte Forschung, im Druck (https://www.jci.org/articles/view/124159; Impact Factor 13,251).
„Über diese interdisziplinäre Herangehensweise, an der neben weiteren Mitarbeitern des Instituts für Humangenetik unter anderem Kollegen des hiesigen Instituts für Neuropathologie beteiligt waren, konnten wir letztlich eine genetisch bedingte neurodegenerative Erkrankung sowie den zugrundeliegenden zellphysiologischen Krankheitsmechanismus beschreiben“, so Prof. Kurth. Inwieweit eine dauerhafte diätetische Umstellung der Ernährung auf Sphingolipid-reiche Kost therapeutisch relevant sein könnte, wollen die Forscher nun testen.