



Kraniofaziale Fehlbildungen
Kraniofaziale Fehlbildungen werden in syndromale und nicht syndromale Anomalien unterteilt. Dabei sind die nicht syndromalen Varianten deutlich häufiger. Die selteneren syndromalen Fehlbildungen gehen meist mit typischen Symptomkomplexen einher (siehe unten). Zu den nicht syndromalen kraniofazialen Fehlbildungen gehören zum Beispiel auch die separat aufgeführten Lippen-, Kiefer- und Gaumenspalten oder Dysgnathien. Bei den so genannten Kraniosynostosen unterscheidet man grundsätzlich die unten aufgeführten Grundformen nach Marchac und Renier, welche in ihrer Ausprägung sehr unterschiedlich ausfallen können. In den meisten Fällen sind Kraniosynostosen vor allem ein ästhetisches Problem, welches sich im Laufe des Wachstumsprozesses häufig weiter verschlechtert. Nur bei stark ausgeprägten Schädeldeformitäten muss mit funktionellen Einschränkungen gerechnet werden.
Schädelformen nach Marchac und Renier (2006):
„Dreieckige“ spitz zulaufende Stirn als Folge einer frühzeitigen Verknöcherung der Sutura metopica. Die Deformität setzt sich bis in Augenregion fort. Typischerweise ist der Augenabstand vermindert.
Das oxyzephale Wachstumsmuster resultiert aus einer gleichzeitigen Verknöcherung von beiden Coronarnähten und zusätzlich weiteren Schädelnähten (z. B. Saggitalnaht). Hier treten im Verlauf auch häufig funktionelle Einschränkungen auf. Typisch für diese Form der Kraniosynostose ist eine hohe Stirn.
Hierbei kommt es zu einer Stirnabflachung auf der betroffenen Seite, welche sich bis in die Schläfenregion fortsetzt und sich während des Wachstums weiter verschlechtert. Ursächlich ist hier eine verfrühte einseitige Verknöcherung der Coronarnaht. Auch bei dieser Form der Kraniosynostose kann es zu einer starken Beeinträchtigung des Mittelgesichts kommen.
Bei dieser Form sind wie beim Oxyzephalus beide Coronarnähte vorzeitig verknöchert. Durch eine gleichzeitige Verknöcherung des Keilbeins ist hier auch der Gesichtsschädel beeinträchtig. Typische Folgen sind ein vergrößerter Augenabstand.
Der so genannte Kahnschädel entsteht durch den vorzeitigen Verschluss der Sagittalnaht. Dabei kommt es bei der Seitenansicht des Kopfes zu einem deutlichen Längenüberschuss bei gleichzeitiger Verschmälerung des Kopfes bei der Aufsicht von oben. Die Stirn ist hier oft schmal.
Beispiele für syndromale kraniofaziale Anomalien sind:
Hierbei handelt sich um eine vererbte Fehlbildung mit vorzeitiger Verknöcherung der Schädeldecke. Es zeigt sich das Bild eines Turm- oder Spitzschädel ohne Hinterkopfausformung, des Weiteren besteht eine Unterentwickelung des Mittelgesichts. Teilweise ist zusätzlich eine Hart- und Weichgaumenspalte vorhanden und Verwachsungen der Finger und Zehen sind möglich.
Diese vererbte Fehlbildung besteht ebenfalls in der vorzeitigen Verknöcherung bestimmter Schädelnähte. So kommt es zu einem typischen Aussehen: kurzer breiter Schädel, vorstehende Augen, evtl. Schielen, zurückliegendes Mittelgesicht, Unterentwicklung des Oberkiefers, scheinbare Vergrößerung des Unterkiefers, auch ein ausgeprägter Fehlbiss und Zahnfehlstellungen sind möglich.
Eine Unterentwicklung der Gesichtsknochen (insbesondere des Jochbeins), fehlgebildete Ohrmuscheln und ein Abfall der Lidspaltenlinie sind kennzeichnend für diese Fehlbildung. Auch können das Unterlid, die Hornhaut und die Regenbogenhaut betroffen sein. Ebenfalls ist eine Beeinträchtigung der Sinnesleistung (Sehen und Hören) möglich, um bleibende Schäden zu vermeiden, sollte dies frühzeitig abgeklärt werden.
Bei diesem Fehlbildungssyndrom kommt es zu einer einseitigen Unterentwicklung. Betroffen sind Ohrmuschel (bis hin zur Nichtanlage), Jochbögen, Augen und Unterkiefer. Fehlbildungen des Mittelohres werden seltener beobachtet. Auch hier sollte eine Abklärung der Sinnesfunktionen (Sehen und Hören) zeitnah erfolgen.
Behandlung
Aufgrund der Vielzahl an verschiedenen Ausprägungsformen der Gesichts- und Schädeldeformitäten ist eine interdisziplinäre Behandlung der kleinen Patienten nötig. Besonders hervorheben möchten wir hier die gute und enge Zusammenarbeit mit unseren Partnern aus der Klinik für Neurochirurgie. Nach Vorstellung in unserer Sprechstunde (Sprechstunde für Lippen-Kiefer-Gaumenspalten und kraniofaziale Fehlbildungen, jeden Donnerstag, Termin nach telefonischer Vereinbarung) wird ein gemeinsames, für jedes Kind individuelles, Behandlungskonzept ausgearbeitet.
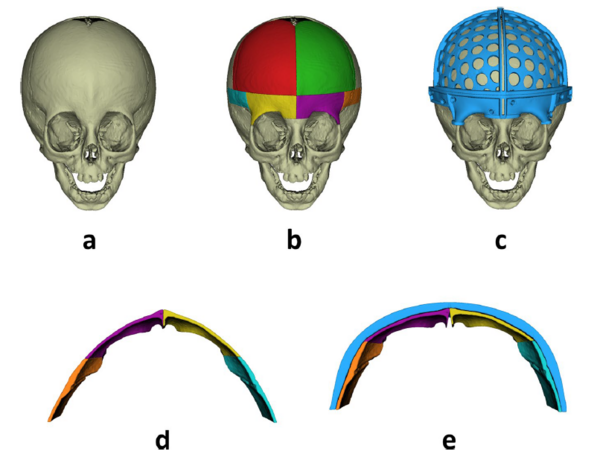
Ist die Indikation für eine Operation gestellt, werden Vorbereitungen für die Operation getroffen. Zur exakten Planung ist eine präoperative Bildgebung erforderlich. Hierfür setzen wir zur Vermeidung einer Röntgenstrahlenexposition vermehrt auf den Einsatz von hochauflösenden Magnetresonanztomographen (MRT). Im Anschluss an diese dreidimensionale Bildgebung erfolgt zusammen mit unseren Kollegen aus der Neurochirurgie und unserem Ingenieur eine detaillierte virtuelle Operationsplanung. Hierdurch kann ein bestmögliches ästhetisches und funktionelles Ergebnis planbar erreicht werden. Die virtuelle Planung wird in der Operation selbst durch so genannte „cutting guides“ zielgenau umgesetzt. Diese Schablonen geben uns nicht nur die geplanten Knochenschnitte vor, sondern ermöglichen uns auch die passgenaue Umsetzung der neu angestrebten Schädelform. Bei der Fixation der verschiedenen knöchernen Fragmente setzen wir ausschließlich auf resorbierbare Materialien. So können unnötige Folgeeingriffe zur Materialentfernung vermieden werden.
In unserer Klinik legen wir besonders Wert auf die umfassende Aufklärung der Eltern des betroffenen Kindes sowie auf ein individuelles Behandlungskonzept, das in Zusammenarbeit mit der Kinderklinik und der neurochirurgischen Klinik erstellt und durchgeführt wird. Deshalb bieten wir Ihnen gerne eine ausführliche Beratung nach telefonischer Anmeldung in unserer Sprechstunde an (Tel.: 0241 80-88230 oder -88231).
Unser interdisziplinäres Behandlungsteam











